 ∷
∷
 ∷
∷
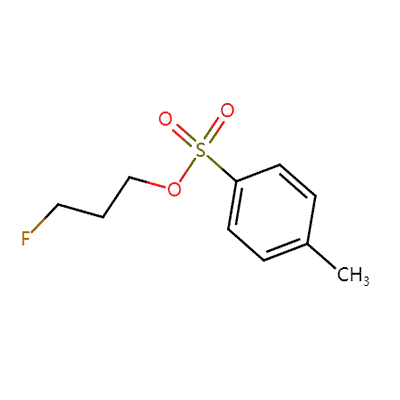
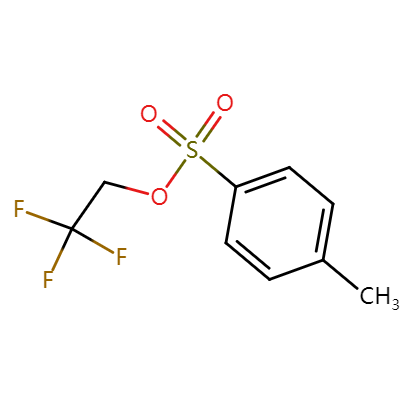
| 3-FLUOROPROPYL P-TOLUENESULFONATE Basic information |
| Product Name: | 3-FLUOROPROPYL P-TOLUENESULFONATE |
| Synonyms: | 3-FLUOROPROPYL P-TOLUENESULFONATE;3-Fluoroprop-1-yl toluene-4-sulphonate;3-Fluoroprop-1-yl 4-methylbenzenesulphonate;3-Fluoroprop-1-yl 4-methylbenzenesulphonate, 3-Fluoroprop-1-yl tosylate;3-Fluoro-1-propanol 1-tosylate;1-Propanol, 3-fluoro-,1-(4-methylbenzenesulfonate) |
| CAS: | 312-68-5 |
| MF: | C10H13FO3S |
| MW: | 232.27 |
| EINECS: | |
| Product Categories: | |
| Mol File: | 312-68-5.mol |
|
|
|
| 3-FLUOROPROPYL P-TOLUENESULFONATE Chemical Properties |
| Boiling point | 160 °C(Press: 2 Torr) |
| density | 1?+-.0.06 g/cm3(Predicted) |
| Safety Information |
|
|
| MSDS Information |
[Tetrahedron, 2019, vol. 75, # 39, art. no. 130529]
[Shengwu Yixue Gongchengxue Zazhi/Journal of Biomedical Engineering, 2017, vol. 34, # 2, p. 253 - 259]
[Journal of labelled compounds and radiopharmaceuticals, 2006, vol. 49, # 14, p. 1247 - 1258]
Synthesis of fluorinated acyclic nucleoside phosphonates with 5-azacytosine base moiety
Abstract
With respect to the strong antiviral activity of (S)-1-[3-hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine various types of its side chain fluorinated analogues were prepared. The title compound, (S)-1-[3-fluoro-2-(phosphonomethoxy)propyl]-5-azacytosine (FPMP-5-azaC) was synthesised by the condensation reaction of (S)-2-[(diisopropoxyphosphoryl)methoxy)-3-fluoropropyl p-toluenesulfonate with a sodium salt of 5-azacytosine followed by separation of appropriate N1 and O2 regioisomers and ester hydrolysis. Transformations of FPMP-5-azaC to its 5,6-dihydro-5-azacytosine counterpart, amino acid phosphoramidate prodrugs and systems with an annelated five-membered imidazole ring, i.e. imidazo [1,2-a][1,3,5]triazine derivatives were also carried out. 1-(2-Phosphonomethoxy-3,3,3-trifluoropropyl)-5-azacytosine was prepared from 5-azacytosine and trifluoromethyloxirane to form 1-(3,3,3-trifluoro-2-hydroxypropyl)-5-azacytosine which was treated with diisopropyl bromomethanephosphonate followed by deprotection of esters. Antiviral activity of all newly prepared compounds was studied. FPMP-5-azaC diisopropyl ester inhibited the replication of herpes viruses with EC50 values that were about three times higher than that of the reference anti-HCMV drug ganciclovir without displaying cytotoxicity.
Primary study on fluro [19F] berberine derivative for human hepatocellular carcinoma targetting in vitro
Abstract
[18F]HX-01, a Fluorine-18 labeled berberine derivative, is a potential positron emission tomography (PET) tumor imaging agent, while [19F]HX-01 is a nonradioactive reference substance with different energy state and has the same physical and chemical properties. In order to collect data for further study of [18F]HX-01 PET imaging of hepatocellular carcinoma in vivo, this study compared the uptake of [19F]HX-01 by human hepatocellular carcinoma and normal hepatocytes in vitro. The target compound, [19F]HX-01, was synthesized in one step using berberrubine and 3- fluoropropyl 4-methylbenzenesulfonate. Cellular uptake and localization of [19F]HX-01 were performed by a fluorescence microscope in human hepatocellular carcinoma HepG2, SMMC-7721 and human normal hepatocyte HL-7702. Cellular proliferation inhibition and cell cytotoxicity assay of the [19F]HX-01 were conducted using cell counting kit-8 (CCK-8) on HepG2, SMMC-7721 and HL-7702 cells. Fluorescent microscopy showed that the combining ability of [19F]HX-01 to the carcinoma SMMC-7721 and HepG2 was higher than that to the normal HL-7702. Cellular proliferation inhibition assay demonstrated that [19F]HX-01 leaded to a dose-dependent inhibition on SMMC-7721, HepG2, and HL-7702 proliferation. Cell cytotoxicity assay presented that the cytotoxicity of [19F]HX-01 to SMMC-7721 and HepG2 was obviously higher than that to HL-7702. This in vitro study showed that [19F]HX-01 had a higher selectivity on human hepatocellular carcinoma cells (SMMC-7721, HepG2) but has less toxicity to normal hepatocytes (HL-7702). This could set up the idea that the radioactive reference substance [18F]HX-01 may be worthy of further development as a potential molecular probe targeting human hepatocellular carcinoma using PET.
Radiosynthesis of 3-(3-[F]fluoropropoxy)-4-(benzyloxy)-N-[(1- dimethylaminocyclopentyl)methyl]-5-methoxybenzamide, a potential PET radiotracer for the glycine transporter GlyT-2
Abstract
The recently described selective and potent GlyT2 antagonist, 4-benzyloxy-3, 5-dimethoxy-N-[(1-dimethylaminocyclopentyl) methyl]benzamide (IC50 = 16 nM) provided an important additional tool to further characterize GlyT2 pharmacology. In order to identify an effective PET radioligand for in vivo assessment of the GlyT-2 transporter, 3-(3-[ F]fluoropropoxy)-4-(benzyloxy)-N-((1-dimethylaminocyclopentyl) methyl)-5-methoxybenzamide ([F]3), a novel analog of 4-benzyloxy-3,5-dimethoxy-N-[(1-dimethylaminocyclopentyl) methyl]benzamide was synthesized using a one-pot, two-step method. The NCA radiofluorination of 1,3-propanediol di-p-tosylate in the presence of K2CO3 and Kryptofix-222 in acetonitrile gave 81% 3-[F]fluoropropyl tosylate, which was subsequently coupled with 4-benzyloxy-3-hydroxy-5-methoxy-N-[(1- dimethylaminocyclopentyl) methyl]-benzamide in the same reaction vessel. Solvent extraction and HPLC (Eclipse XDB-C8 column, 80/20/0.1 MeOH/H 2O/Et3N, 3.0 ml/min) gave [F]3 in 98.5% radiochemical purity. The radiochemical yield was determined to be 14.0-16.2% at EOS, and the specific activity was 1462 ± 342 GBq/μmol. The time of synthesis and purification was 128 min. The final product was prepared as a sterile saline solution suitable for in vivo use. Copyright 2006 John Wiley & Sons, Ltd.
Contact: Mr Hsu
Phone: +86(0)1815701-8567
Tel: +86(0)570780-5618
Email: sales@fluorio.com
Add: No.5,Longfei Road,Longyou Eco-Tech Development Zone, Quzhou, Zhejiang, China